Inference of transcription factor binding and motif finding
Accurate inference of coding sequence annotation using Ribosome profiling data
Genome-wide association analysis of complex traits
Bayesian Co-estimation of Alignment and Tree
Multi-scale analyses of functional data, including phenotypes arising from high-throughput sequencing assays
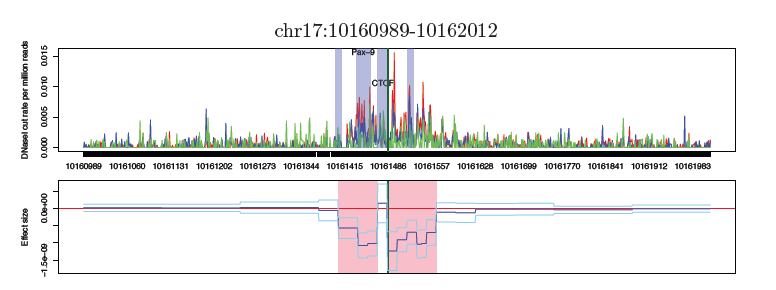
Identification of differences between multiple groups in molecular and cellular phenotypes measured by high-throughput sequencing assays is frequently encountered in genomics applications. For example, common problems include eQTL mapping using RNA-seq and detecting differences in chromatin accessibility across tissues using DNase-seq or ATAC-seq. Those high-throughput sequencing data provide high-resolution measurements on how traits vary along the whole genome in each sample. However, typical analyses fail to exploit the full potential of these high-resolution measurements, instead aggregating the data at coarser resolutions, such as genes, or windows of fixed length. We developed wavelet-based methods that more fully exploit the high-resolution data, and produced a software package WaveQTL. Applying these methods to identify genetic variants associated with chromatin accessibility (dsQTLs) we identified 50% more dsQTLs than a typical window-based analysis in Degner et al. (2012) and, importantly, identified dsQTL with different biological interpretation than those identified in the window-based analysis.
Motivated by this, we have developed statistical methods that model the count nature of the sequence data directly using multi-scale models, and produced a software package multiseq. As expected, multiseq outperforms WaveQTL in smaller sample sizes. Even with larger sample sizes (e.g., 70), multiseq outperforms WaveQTL unless library read depths are very high. Moreover, we applied these two multi-scale methods and a window-based approach, DESeq, to ATAC-seq data measured on Copper-treated samples and control samples (3 vs 3), and we found that multiseq detected substantially more differences in chromatin accessibility between two conditions than WaveQTL and DESeq.
We are currently working on multiple projects to improve these two approaches (e.g., modeling dependencies of wavelet coefficients using hidden Markov tree models). While we developed these methods with specific applications to sequencing data in mind, these methods have natural applications for analysis of many complex functional phenotypes.
- Shim and Stephens (2014) [link][software WaveQTL][supplementary materials][supplementary figures]
- Shim et al. (In prep.) [software multiseq][github for analysis]
- Shim*, Zuk* et al. (In progress)
Inference of transcription factor binding and motif finding
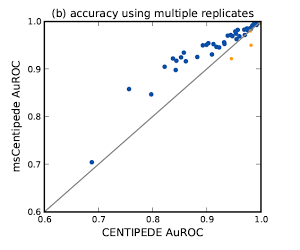
Understanding global gene regulation critically depends on accurate annotation of regulatory elements that are functional in a given cell type. CENTIPEDE, a powerful, probabilistic framework for identifying transcription factor binding sites from tissue-specific DNase I cleavage patterns and genomic sequence content, leverages the hypersensitivity of factor-bound chromatin and the information in the DNase I spatial cleavage profile characteristic of each DNA binding protein to accurately infer functional factor binding sites. However, the model for the spatial profile in this framework underestimates the substantial variation in the profiles across genomic locations and across replicate measurements of chromatin accessibility. We use hierarchical multi-scale models for Poisson processes to better model the additional variation. Using DNase-seq measurements assayed in a lymphoblastoid cell line, we demonstrate the improved area under the Receiver Operating Characteristic of this model for several transcription factors by comparing against the Chip-Seq peaks for those factors.
Identifying motifs of transcription factors is usually addressed in a two-stage process based on ChIP-seq data. First, candidate bound sequences that are in the order of 500-1000 bp are identified from the ChIP-seq data. Then, motif searches are performed among these sequences. These 2 steps are typically carried out in a disconnected fashion in the sense that the quantitative nature of the ChIP-seq information is ignored in the second step. We developed a conditional two-component mixture model that adaptively integrates the quantitative information into motif finding. We found that the proposed method has superior sensitivity and specificity especially when the motif of interest has low abundance among the candidate bound sequences and/or low information content.
- Raj*, Shim* et al. (2015) [link][software msCentipede]
- Shim and Keles (2007) [link][software SUCcESS]
Accurate inference of coding sequence annotation using ribosome profiling data
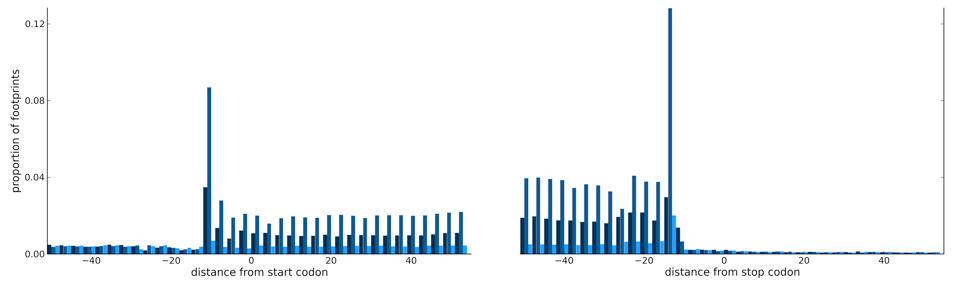
Recent increase in biological data acquisition has significantly improved our annotation of regulatory elements. In contrast, current annotations of the coding sequence (CDS) still largely rely on databases of cDNA clones and peptide sequences (over decades of data accumulation), gene prediction programs and evolutionary conservation. These measures, for the most part, do not directly reflect translation events but are distant surrogates. Recently, a genomics technique has been developed for profiling ribosome occupancy using next generation sequencing platforms. This development makes it possible to measure translation events directly at subcodon precision across the (mappable) genome. We developed a hidden Markov model, riboHMM, to use the ribosome profiling data (and, optionally, the current information on CDS annotation) for accurate inference of CDS annotation.
- Raj*, Wang*, Shim*, et al. [bioRxiv][software riboHMM]
Genome-wide association analysis of complex traits
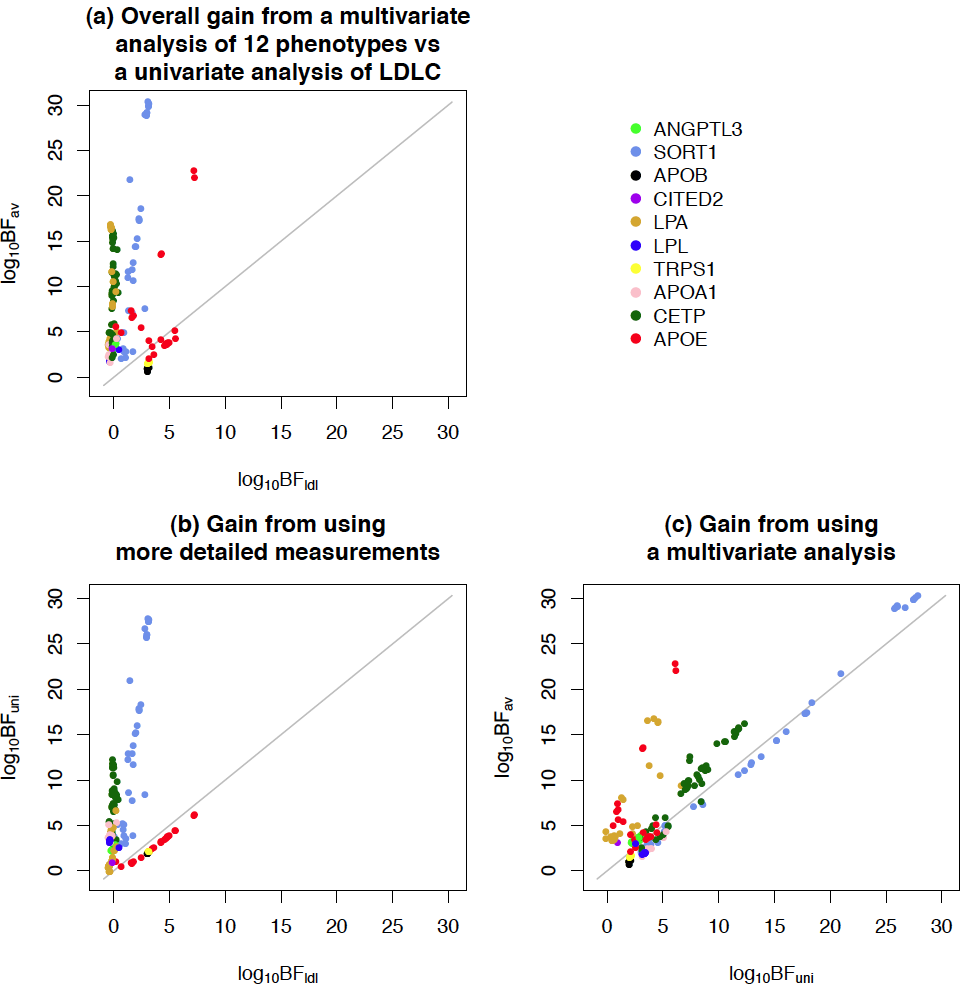
We conducted genetic association analyses of 10 low density lipoprotein (LDL) subfractions, and their response to statin treatment, in 1868 individuals of European ancestry. These phenotypic measurements offer higher resolution information on LDLs than available previously.Unlike most previous analyses of multiple related measurements, we analyzed the subfractions jointly, rather than one at a time. Our results demonstrate that joint analyses can considerably increase power to detect associations compared with conventional univariate analyses. We found multiple loci that contain variants only very weakly associated with total LDL-cholesterol, but are very strongly associated with one or several individual LDL subfractions. The most interesting of the associations are the two independently-associated SNPs in CETP showing strikingly similar patterns of association with combinations of subfractions both in our original data and in a replication cohort.
- Shim et al. (2015) [link][software mvBIMBAM]
Bayesian Co-estimation of Alignment and Tree
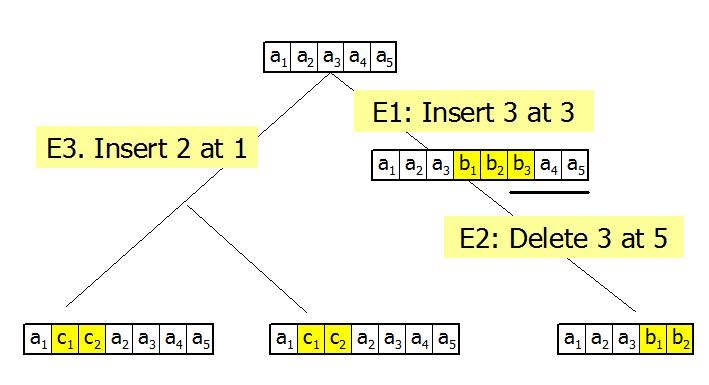
We developed a Bayesian approach for co-estimating phylogeny and sequence alignment. Our method for joint estimation improves estimates from the traditional sequential approach by accounting for uncertainty in the alignment in phylogenetic inferences. Unlike alternative models for joint estimation of alignment and phylogeny, our insertion and deletion (indel) model allows arbitrary-length overlapping indel events and a general distribution for indel fragment size. In addition, we explicitly model a completely history of indel events on the tree. Therefore, our approach enables us to infer more information about the indel process.
- Shim and Larget, (Under review) [pdf][supplementary materials][software BayesCAT]
